|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
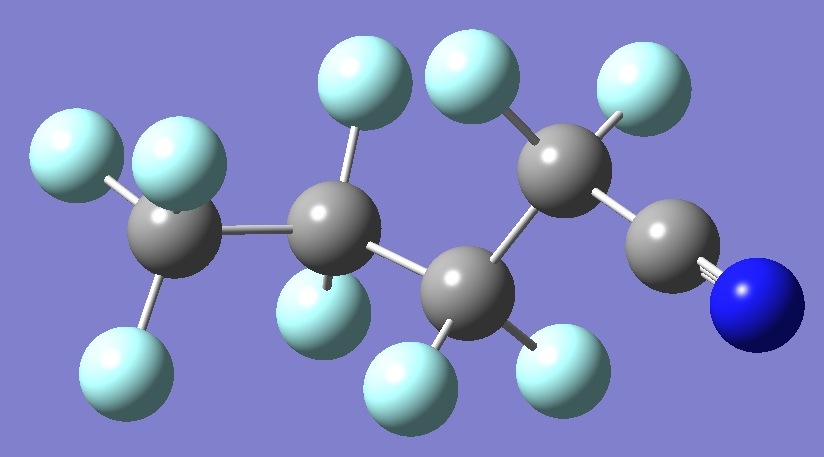
CF3(CF2)2CF2CN
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nitrogen |
|
|
|
Nuclear
Quadrupole Coupling Constants |
|
|
in n-Perfluorobutyl
Cyanide anti-gauche
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Calculation of the nitrogen nqcc's in n-perfluorobutyl cyanide was made
on structures given by
B3P86/6-31G(2d) optimization (ropt), and on this same
structure but with empirically derived
equilibrium CF, C-C, and CN bond lengths
(~ re).
These calculated nqcc's are given in Table 1. Structure
parameters are
given in
Table 2, rotational constants and electric dipole moments in Table 3. |
|
|
|
|
|
|
|
|
|
|
|
|
In Table 1,
subscripts a,b,c refer to
principal axes of the inertia tensor; subscripts x,y,z to principal
axes
of the nqcc tensor. Øz,CN (degrees) is the
angle between the principal z-axis of the nqcc tensor and the CN bond
axis. ETA = (Xxx - Xyy)/Xzz. |
|
|
RSD is the calibration residual
standard deviation of
the B3PW91/6-311+G(df,pd) model for calculation of the nitrogen nqcc's,
which may be taken as an estimate of the uncertainty in the calculated
nqcc's (unertainties in the structures not-with-standing). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Table 1. 14N nqcc's
in n-perfluorobutyl
cyanide, AG (MHz).
Calculation was made on ropt and ~ re structures
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
ropt |
|
~ re |
|
Expt. |
|
| |
|
|
|
|
|
|
|
|
|
Xaa |
- |
2.548 |
- |
2.542 |
|
|
|
|
Xbb |
|
1.830 |
|
1.823 |
|
|
|
|
Xcc |
|
0.718 |
|
0.719 |
|
|
|
|
|Xab| |
|
1.608 * |
|
1.610 * |
|
|
|
|
|Xac| |
|
2.865 |
|
2.858 |
|
|
|
|
|Xbc| |
|
0.985 |
|
0.985 |
|
|
|
|
|
|
|
|
|
|
|
|
|
RSD |
|
0.030 (1.3 %) |
0.030 (1.3 %) |
|
|
|
|
|
|
|
|
|
|
|
|
Xxx |
|
2.334 |
|
2.329 |
|
|
|
|
Xyy |
|
2.419 |
|
2.413 |
|
|
|
|
Xzz |
- |
4.753 |
- |
4.742 |
|
|
|
|
ETA |
|
0.0179 |
|
0.0177 |
|
|
|
|
Øz,CN |
|
0.39 |
|
0.37 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* The algebraic sign of the product XabXacXbc is
negative. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
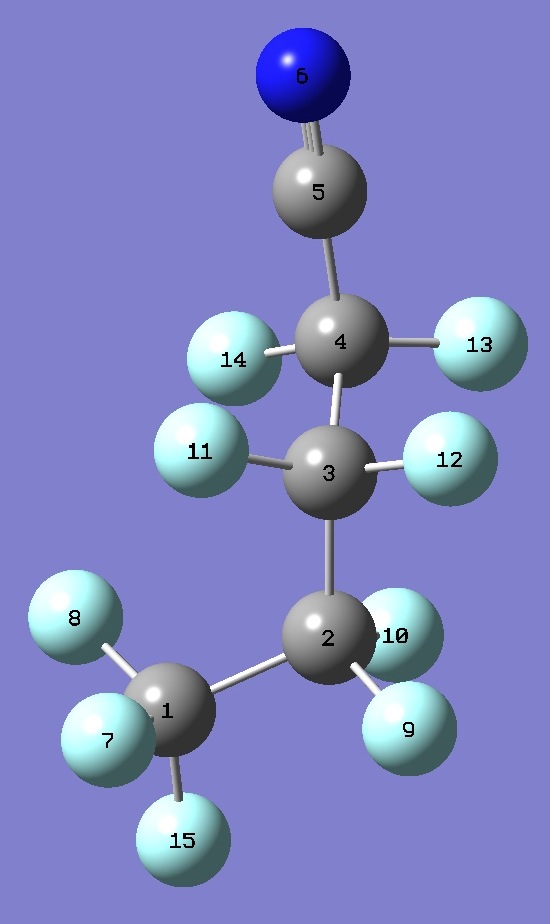
| Table 2.
n-Perfluorobutyl cyanide, AG.
Selected structure parameters, ropt and ~
re (Å and degrees). Complete
structures are given here in
Z-matrix format. |
| |
|
|
|
|
|
|
|
|
ropt |
|
~ re |
|
|
|
|
|
| C(1)C(2) |
|
1.5468 |
|
1.5455 |
| C(2)C(3) |
|
1.5624 |
|
1.5604 |
| C(3)C(4) |
|
1.5504 |
|
1.5490 |
| C(4)C(5) |
|
1.4792 |
|
1.5808 |
| C(5)N |
|
1.1534 |
|
1.1544 |
| C(1)C(2)C(3) |
|
115.02 |
|
115.02 |
| C(2)C(3)C(4) |
|
115.00 |
|
115.00 |
| C(3)C(4)C(5) |
|
109.94 |
|
109.94 |
| C(4)C(5)N |
|
179.72 |
|
179.72 |
| C(1)C(2)C(3)C(4) |
|
- 98.03 |
|
- 98.03 |
| C(2)C(3)C(4)C(5) |
|
172.11 |
|
172.11 |
| C(3)C(4)C(5)N |
|
42.62 |
|
42.62 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
| Table 3.
n-Perfluorobutyl Cyanide, AG. Rotational
Constants (MHz) and Dipole Moments (D). |
|
|
|
|
|
|
|
ropt |
~ re |
Expt. |
|
|
|
|
|
|
A |
1074.1 |
1070.3 |
|
|
B |
470.4 |
470.2 |
|
|
C |
463.0 |
462.9 |
|
|
|
|
|
|
|
|µa| |
1.24 |
1.22 |
|
|
|µb| |
0.32 |
0.31 |
|
|
|µc| |
0.60 |
0.59 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Order of the energies of the several
conformers of n-perfluorobutyl cyanide is Etransoid(0)
< EGA(0.54) < EAA(0.92)
< EAG(6.95
kJ/mol). These are energies calculated at the B3PW91/6-311+G(df)
level of theory on the B3P86/6-31G(2d) optimized structures. |
|
|
The AA (all trans, Cs)
conformer, with one imaginary frequency, is a saddle point connecting
two lower energy minima [1]. It is not, itself, a local minimum. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[1] J.B.Foresman and AE.Frisch,
Exploring Chemistry with Electronic Structure Methods, 2nd ed.,
Gaussian, Inc. Pittsburgh, PA 1995-96. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
n-Butyl
Cyanide AG |
|
|
|
n-Butyl
Cyanide GA |
n-Perfluorobutyl
Cyanide, GA |
|
|
|
n-Butyl
Cyanide AA |
n-Perfluorobutyl
Cyanide, transoid |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table of Contents |
|
|
|
|
|
Molecules/Nitrogen |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C4F9CN_AG.html |
|
|
|
|
|
|
Last
Modified 3 June 2010 |
|
|
|
|
|
|
|
|
|
|