|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(CF3)2C=NH |
|
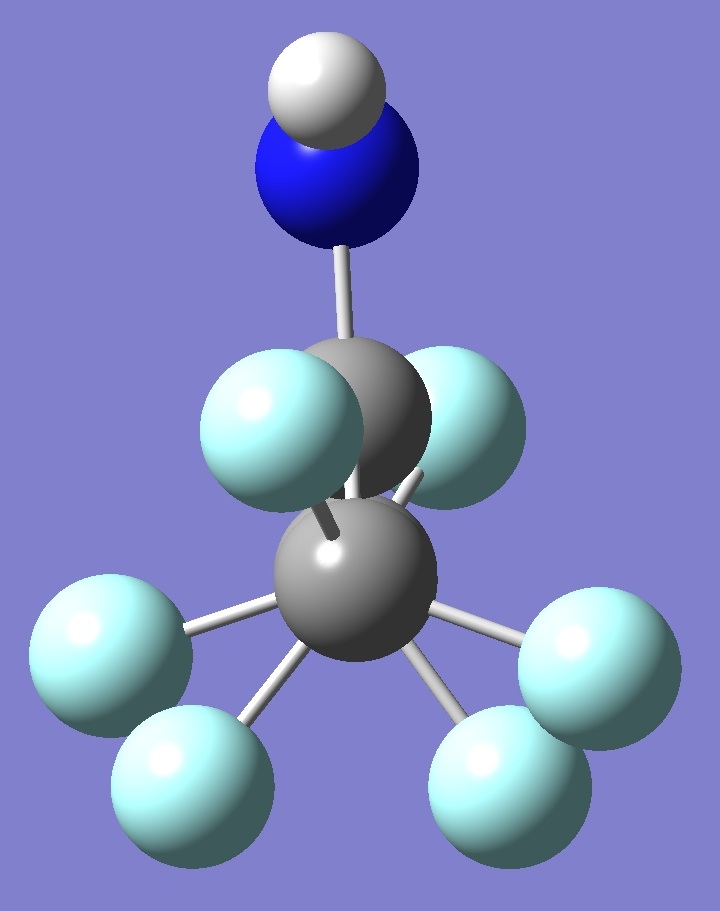 |
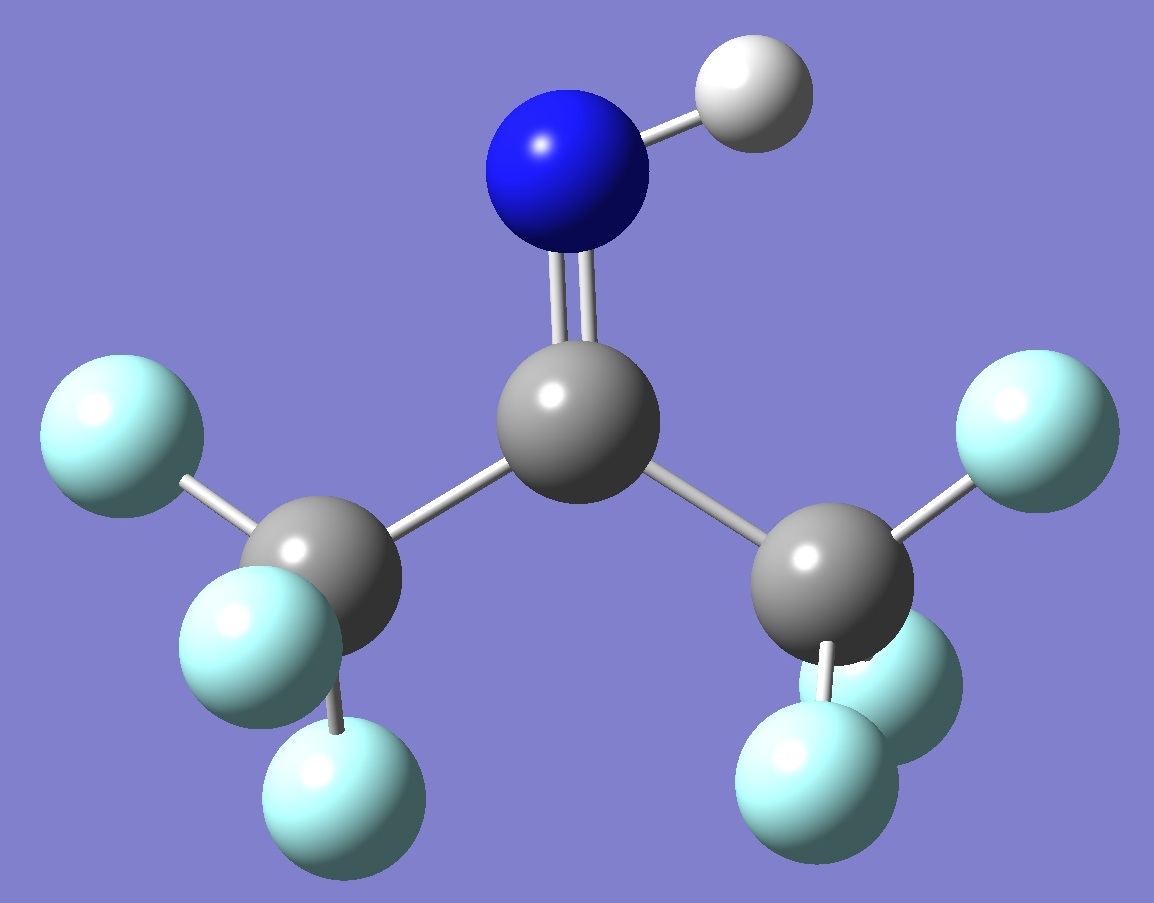 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nitrogen
|
|
|
Nuclear
Quadrupole Coupling Constants |
|
|
in
transoidal
Hexafluoroacetone imine
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nitrogen nqcc's in hexafluoroacetone
imine were
determined by Grubbs II et al. [1]. |
|
|
|
|
|
|
|
|
|
|
|
|
Calculation of the 14N
nqcc tensors was made here on molecular structures derived by
MP2/6-311+G(d,p), MP2/6-311+G(df,pd), and MP2/6-311+G(3df,3pd)
optimizations. Calculated and experimental nqcc's are compared in
Table 1. Structure parameters are given in Table 2, rotational
constants and dipole moment components in Table 3. |
|
|
|
|
|
|
|
|
|
|
|
|
In Table 1, subscripts a,b,c
refer to the
principal axes of the inertia tensor; x,y,z to the principal axes
of the nqcc tensor. Øz,bi (degrees) is the
angle between the z-principal axis and the bisector or the C=NH angle.
ETA = (Xxx - Xyy)/Xzz. |
|
|
RMS is the root mean square
difference between calculated and experimental diagonal nqcc's
(percentage of the average of the magnitudes of the experimental
nqcc's). RSD is the calibration residual standard deviation of
the B3PW91/6-311+G(df,pd) model for calculation of nitrogen nqcc's. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
Table 1. 14N nqcc's in (CF3)2C=NH
(MHz). Calculation was made on (1) MP2/6-311+G(d,p), (2)
MP2/6-311+G(df,pd), and (3) MP2/6-311+G(3df,3pd)
optimized molecular structures. |
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Calc. (1)
|
|
Calc. (2) |
|
Calc. (3) |
|
Expt. [1] |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Xaa |
- |
3.101 |
- |
3.044 |
- |
3.083 |
- |
3.050(17) |
|
|
Xbb |
- |
0.980 |
- |
1.015 |
- |
1.026 |
- |
1.008(22) |
|
|
Xcc |
|
4.081 |
|
4.059 |
|
4.109 |
|
4.058(14) |
|
|
Xab |
|
2.420 |
|
2.431 |
|
2.461 |
|
|
|
|
Xac |
- |
0.242 |
- |
0.238 |
- |
0.228 |
|
|
|
|
Xbc |
|
0.075 |
|
0.074 |
|
0.069 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
RMS |
|
0.036 (1.3 %) |
0.005 (0.20 %) |
0.036 (1.3 %) |
|
|
|
RSD |
|
0.030 (1.3 %) |
0.030 (1.3 %) |
0.030 (1.3 %) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Xxx |
|
0.600 |
|
0.604 |
|
0.612 |
|
|
|
|
Xyy |
|
4.090 |
|
4.066 |
|
4.116 |
|
|
|
|
Xzz |
- |
4.690 |
- |
4.670 |
- |
4.728 |
|
|
|
|
ETA |
|
0.744 |
|
0.741 |
|
0.741 |
|
|
|
|
Øz,bi * |
|
1.12 |
|
1.57 |
|
1.52 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Angle between the z-principal axis
and the bisector or the C=NH angle. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Table 2.
(CF3)2C=NH. Selected structure
parameters (Å and
degrees). Complete structures are given here
in Z-matrix format. |
| |
|
|
|
|
|
|
|
ropt (1) =
MP2/6-311+G(d,p) optimization. |
|
|
ropt (2) =
MP2/6-311+G(df,pd) optimization. |
|
|
ropt (3) =
MP2/6-311+G(3df,3pd optimization. |
| |
|
|
|
|
|
|
|
ropt (1) |
ropt (2) |
ropt (3) |
|
|
|
|
|
|
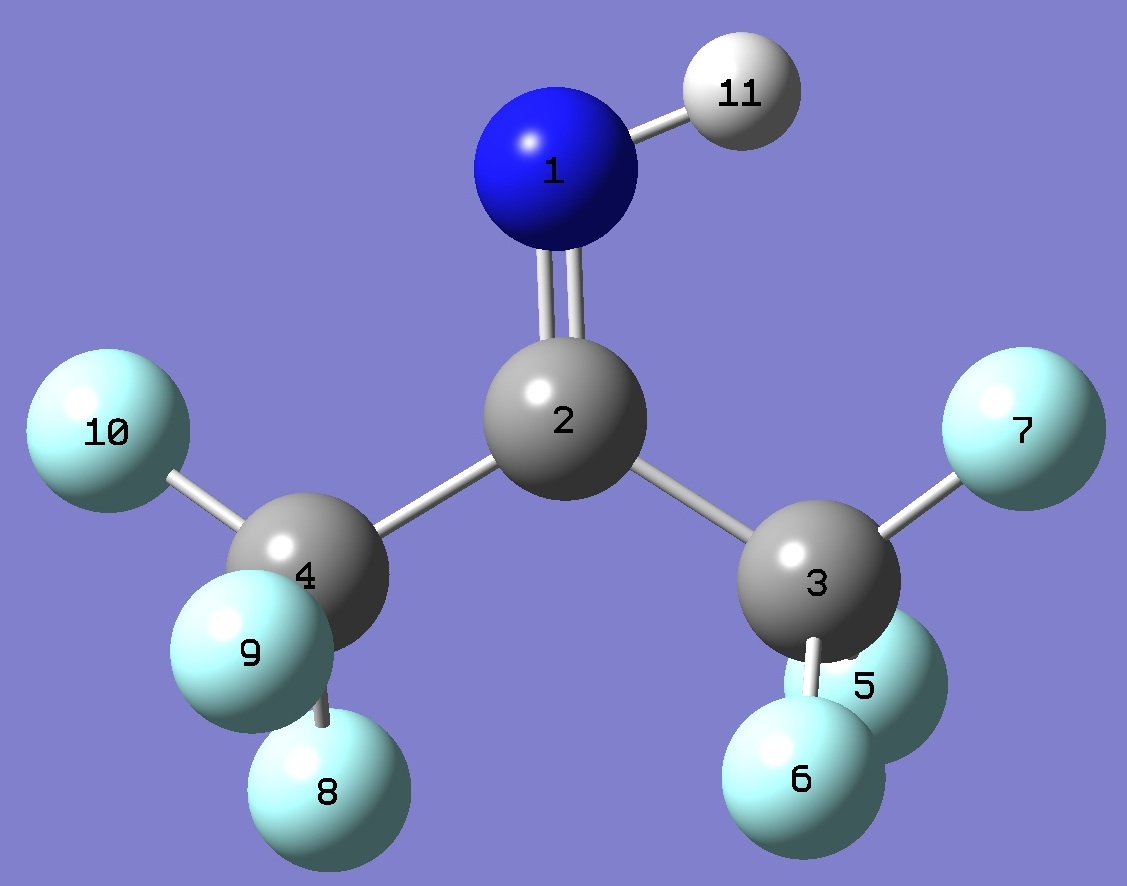 |
C(2)C(4) |
1.5254 |
1.5267 |
1.5237 |
| C(2)C(3) |
1.5275 |
1.5289 |
1.5256 |
| C(2)N |
1.2712 |
1.2672 |
1.2646 |
| NH |
1.0242 |
1.0229 |
1.0207 |
| C(4)C(2)C(3) |
116.36 |
116.32 |
116.18 |
| C(4)C(2)N |
118.60 |
118.68 |
118.99 |
| C(3)C(2)N |
125.02 |
124.99 |
124.82 |
| C(2)NH |
110.05 |
110.01 |
110.50 |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
| Table 3. (CF3)2C=NH.
Rotational constants (MHz) and dipole moment components (D). |
| |
|
|
|
|
|
|
ropt (1) =
MP2/6-311+G(d,p) optimization. |
|
ropt (2) =
MP2/6-311+G(df,pd) optimization. |
|
ropt (3) =
MP2/6-311+G(3df,3pd) optimization. |
|
|
|
|
|
|
|
|
Calc. ropt (1) |
Calc. ropt (2) |
Calc. ropt (3) |
Expt. [1] |
|
|
|
|
|
|
|
A |
2159.1 |
2183.1 |
2175.4 |
2143.0309(12) |
|
B |
1045.4 |
1052.3 |
1052.6 |
1044.50289(24) |
|
C |
938.0 |
943.7 |
944.2 |
934.5118(11) |
|
|
|
|
|
|
|
|µa| |
1.45 |
1.46 |
1.44 |
|
|
|µb| |
0.92 |
0.84 |
0.89 |
|
|
|µc| |
0.01 |
0.01 |
0.01 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Note: The Cs
conformer of this molecule, with one imaginary frequency (as calculated
at the MP2/6-311+G(d,p) level of theory), is a first order transition
state connecting two lower energy conformers, presumely this transoidal
conformer and its mirror image conformer. E(Cs) >
E(transoid) by about 2.6 kJ/mole. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[1] G.S.Grubbs II, C.T.Dewberry,
B.E.Long, W.C.Pringle, and S.A.Cooke, 66th International Symposium on
Molecular Spectroscopy, Ohio State University, 2011, Abstract WH14. |
|
|
|
|
|
|
|
|
|
|
|
|
D.A.Obenchain, D.J.Frohman, G.S.Grubbs II,
B.E.Long, W.C.Pringle, S.E.Novick, and S.A.Cooke, 67th International Symposium on
Molecular Spectroscopy, Ohio State University, 2012, Abstract TC04. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H2C=NH |
F2C=NH |
F2C=NF |
trans-syn-Propenimine |
|
t-Ethanimine |
c-Ethanimine |
H2C=NOH |
trans-anti-Propenimine |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table of Contents |
|
|
|
|
|
Molecules/Nitrogen |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CF32CNH.html |
|
|
|
|
|
|
Last
Modified 7 July 2011 |
|
|
|
|
|
|
|
|
|
|