| |
||||||||
| Table 1. D nqcc's in H-CC-C(=O)OD (kHz). Calculation was made on the (1) MP2/6-311+G(3df,3pd) and (2) MP2/aug-cc-pVTZ ropt molecular structures. | ||||||||
| |
||||||||
| Calc (1) | Calc (2) |
Expt [1] | ||||||
| |
||||||||
| Xaa | 260.8 | 255.3 | 256.5(33) * |
|||||
| Xbb | - |
110.7 | - |
108.3 | - |
112.6(45) * |
||
| Xcc | - |
150.1 | - |
147.0 | - |
143.9(45) * |
||
| |Xab| | 61.5 |
59.7 |
||||||
| RMS | 4.4 (2.6 %) |
3.1 (1.8 %) |
||||||
| RSD | 1.1 (0.86 %) |
1.1 (0.86 %) | ||||||
| Xxx | - |
120.6 | - |
117.8 | ||||
| Xyy | - |
150.1 | - |
147.0 | ||||
| Xzz | 270.7 | 264.9 | ||||||
| ETA | 0.109 | 0.110 | ||||||
| Øz,a |
9.17 |
9.10 |
||||||
| Øa,OD | 10.62 |
10.56 |
||||||
| Øz,OD | 1.45 |
1.46 |
||||||
| |
||||||||
| |
||||||||
| Table 2. D nqcc's in D-CC-C(=O)OH (kHz). Calculation was made on the (1) MP2/6-311+G(3df,3pd) and (2) MP2/aug-cc-pVTZ ropt molecular structures. | ||||||||
| |
||||||||
| Calc (1) | Calc (2) |
Expt [1] | ||||||
| |
||||||||
| Xaa | 212.9 | 212.6 | 205.3(27) * |
|||||
| Xbb | - |
108.7 | - |
108.5 | ||||
| Xcc | - |
104.2 | - |
104.1 | ||||
| |Xab| | 5.4 |
5.7 |
||||||
| RMS | 7.6 (3.7 %) |
7.3 (3.6 %) |
||||||
| RSD | 1.1 (0.86 %) |
1.1 (0.86 %) | ||||||
| Xxx | - |
108.8 | - |
108.6 | ||||
| Xyy | - |
104.2 | - |
104.1 | ||||
| Xzz | 213.0 | 212.7 | ||||||
| ETA | 0.0213 | 0.0214 | ||||||
| Øz,a |
0.96 |
1.014 |
||||||
| Øa,CD | 0.96 |
1.017 |
||||||
| Øz,CD | 0.00 |
0.003 |
||||||
| |
||||||||
| Table 3. Propiolic Acid molecular structure parameters, ropt(1) = MP2/6-311+G(3df,3pd) optimization and ropt(2) = MP2/aug-cc-pVTZ optimization (Å and degrees). | ||
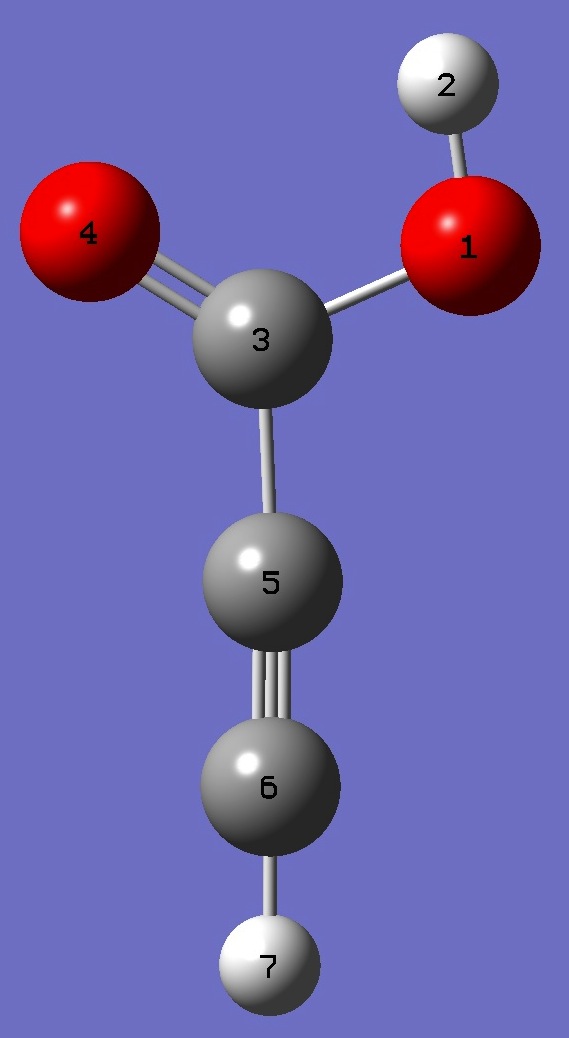 |
O H,1,B1 C,1,B2,2,A1 O,3,B3,1,A2,2,D1,0 C,3,B4,1,A3,4,D2,0 C,5,B5,2,A4,4,D3,0 H,6,B6,4,A5,2,D4,0 |
|
| ropt(1) | ropt(2) | |
| |
||
| B1=0.96815607 B2=1.34618353 B3=1.20674381 B4=1.44665384 B5=1.21305172 B6=1.06234116 A1=105.75550831 A2=124.15443066 A3=111.39565131 A4=161.14605072 A5=161.75136421 D1=0. D2=180. D3=180. D4=180. |
B1=0.97119556 B2=1.34964818 B3=1.21003443 B4=1.44601681 B5=1.21427027 B6=1.0625963 A1=105.73105785 A2=124.12112095 A3=111.3002228 A4=161.1340831 A5=161.82450926 D1=0. D2=180. D3=180. D4=180. |
|
| Table 4. Propiolic Acid rotational constants (MHz). ropt(1) = MP2/6-311+G(3df,3pd), ropt(2) = MP2/aug-cc-pVTZ optimized structures. | |||||
| ropt(1) | ropt(2) | Expt [1] | |||
| HCCC(=O)OD | A |
11848.4 |
11788.7 |
11858.44934(132) |
|
| B |
4003.1 |
3998.0 |
4015.71252(41) |
||
| C |
2992.2 |
2985.5 |
2995.59587(48) |
||
| |
|||||
| DCCC(=O)OH |
A |
12100.2 |
12040.2 |
12110.01758(217) |
|
| B |
3805.7 |
3801.0 |
3819.67859(110) |
||
| C |
2895.1 |
2889.0 |
2899.60416(55) |
||