|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH2·NH·CH(CN)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nitrogen
|
|
|
Nuclear
Quadrupole Coupling Constants |
|
|
|
in 2-Cyanoaziridine |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amino and cyano nitrogen nqcc's in the cis conformer of 2-cyanoaziridine have been determined by Brown et al. [1]. |
|
|
|
|
|
|
|
|
|
|
|
|
Calculation of the nqcc's was
made here on a molecular structure given by MP2/6-311+G(3d,3p)
optimization, with corrected MP2/6-311+G(d,p) optimized CN bond length.
Calculated nqcc's are
compared in Tables 1 and 2 with the experimental nqcc's.
Structure parameters are given in Table 3, atomic coordinates in
Table 4, and rotational constants in Table 5. |
|
|
|
|
|
|
|
|
|
|
|
|
In Tables 1 and 2, subscripts a,b,c refer to the principal axes of the inertia
tensor, subscripts x,y,z to the principal axes of the nqcc tensor. Ø (degrees)
is the angle between its subscripted parameters. ETA = (Xxx
- Xyy)/Xzz. |
|
|
RMS is the root mean square
difference between calculated and experimental diagonal nqcc's (percentage of
average experimental nqcc). RSD is the residual standard deviation
of calibration of the B3PW91/6-311+G(df,pd) model for calculation of
the nqcc's. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
Table 1.
Amino Nitrogen nqcc's in 2-Cyanoaziridine (MHz). |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Calc. |
|
Expt. [1] |
|
| |
|
|
|
|
|
|
|
|
14N |
Xaa |
- |
1.214 |
- |
1.249(14) |
|
|
|
Xbb |
|
1.410 |
|
1.407(12) |
|
|
|
Xcc |
- |
0.197 |
- |
0.158 * |
|
|
|
Xab** |
|
0.786 |
|
|
|
|
|
Xac** |
|
2.456 |
|
|
|
|
|
Xbc** |
- |
2.145 |
|
|
|
|
|
|
|
|
|
|
|
|
|
RMS |
|
0.030 (3.2 %) |
|
|
|
|
|
RSD |
|
0.030 (1.3 %) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Xxx |
|
0.879 |
|
|
|
|
|
Xyy |
|
3.085 |
|
|
|
|
|
Xzz |
- |
3.964 |
|
|
|
|
|
ETA |
|
0.557 |
|
|
|
|
|
Øz,NH |
|
134.42 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
* Derived here from zero trace condition. |
|
|
** Algebraic signs of the off-diagonal
components of the nqcc tensor depend on the molecular orientation with
respect to the positive/negative sense (polarity) of a,b,c-coordinates.
The algebraic sign of the product XabXacXbc, which is negative in this case, in independent of polarity. |
|
|
The algebraic signs given here correspond to the molecular orientation with respect to a,b,c coordinates given in Table 3. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
Table 2. Cyano Nitrogen nqcc's in 2-Cyanoaziridine (MHz). |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Calc. |
|
Expt. [1] |
|
| |
|
|
|
|
|
|
|
|
14N |
Xaa |
- |
3.499 |
- |
3.547(6) |
|
|
|
Xbb |
|
1.794 |
|
1.865(8) |
|
|
|
Xcc |
|
1.705 |
|
1.682 * |
|
|
|
Xab** |
- |
0.097 |
|
|
|
|
|
Xac** |
|
2.127 |
|
|
|
|
|
Xbc** |
- |
0.120 |
|
|
|
|
|
|
|
|
|
|
|
|
|
RMS |
|
0.051 (2.2 %) |
|
|
|
|
|
RSD |
|
0.030 (1.3 %) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Xxx |
|
1.764 |
|
|
|
|
|
Xyy |
|
2.494 |
|
|
|
|
|
Xzz |
- |
4.258 |
|
|
|
|
|
ETA |
|
0.172 |
|
|
|
|
|
Øz,CN |
|
0.23 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Derived here from zero trace condition.
|
|
|
** Algebraic signs of the off-diagonal
components of the nqcc tensor depend on the molecular orientation with
respect to the positive/negative sense (polarity) of a,b,c-coordinates.
The algebraic sign of the product XabXacXbc, which is positive in this case, in independent of polarity. |
|
|
The algebraic signs given here correspond to the molecular orientation with respect to a,b,c coordinates given in Table 3. |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
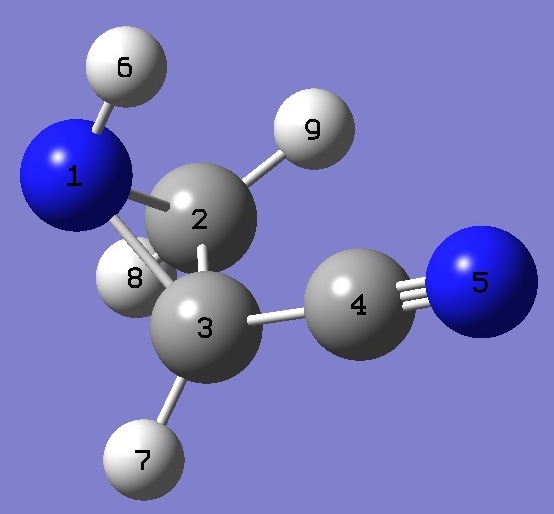
| Table 3. 2-Cyanoaziridine. Heavy atom structure parameters (Å
and degrees). MP2/6-311+G(3d,3p) optimized structure. Complete structure is given here in Z-matrix format. |
| |
|
|
|
|
|
N(1)H(6) |
1.0157 |
|
N(1)C(2) |
1.4714 |
|
C(2)C(3) |
1.4892 |
|
C(3)N(1) |
1.4799 |
|
C(3)C(4) |
1.4423 |
|
C(4)N(5) |
1.1572 * |
|
C(3)N(1)H(6) |
108.76 |
|
N(1)C(2)C(3) |
59.98 |
|
C(2)C(3)N(1) |
59.41 |
|
C(3)N(1)C(2) |
60.61 |
|
C(2)C(3)C(4) |
118.06 |
|
|
C(3)C(4)N(5) |
178.51 |
|
|
|
|
|
* Corrected MP2/6-311+G(d,p) optimized CN bond length [2]. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Table 4. 2-Cyanoaziridine. Atomic coordinates. |
| |
|
|
|
|
|
|
|
|
|
|
a (Å) |
|
b (Å) |
|
c (Å) |
|
|
|
|
|
|
|
|
|
N(1) |
- |
1.3144 |
|
0.7501 |
- |
0.1777 |
|
C(2) |
- |
1.3313 |
- |
0.7201 |
- |
0.2350 |
|
C(3) |
- |
0.3156 |
- |
0.1851 |
|
0.5980 |
|
C(4) |
|
1.0537 |
- |
0.0227 |
|
0.1448 |
|
N(5) |
|
2.1442 |
- |
0.0086 |
- |
0.2420 |
|
H(6) |
- |
0.8680 |
|
1.1150 |
- |
1.0140 |
|
H(7) |
- |
0.4486 |
|
0.0325 |
|
1.6678 |
|
H(8) |
- |
2.1710 |
- |
1.1784 |
|
0.2638 |
|
H(9) |
- |
0.9788 |
- |
1.206494 |
- |
1.1326 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
| Table 5.
2-Cyanoaziridine. Rotational Constants (MHz). |
| |
|
|
|
|
|
Calc. ropt |
Expt. [1] |
|
|
|
|
|
A |
16 900.3 |
16 877.718(32) |
|
B |
3 541.3 |
3 528.931(4) |
|
C |
3 385.6 |
3 373.065(4) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[1] R.D.Brown, P.D.Godfrey, and A.L.Ottrey, J.Mol.Spectrosc. 82,73(1980). |
|
|
[2] J.Demaison, J.Cosléou, R.Bocquet,
and A.G.Lesarri, J.Mol.Spectrosc. 167,400(1994). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3CN |
CH3CH2CN |
|
|
Aziridine (Ethylenimine) |
c-Ethanimine |
|
|
CH2NH |
t-Ethanimine |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table of Contents |
|
|
|
|
|
Molecules/Nitrogen |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2CNAziridine.html |
|
|
|
|
|
|
Last
Modified 15 Sept 2006 |
|
|
|
|
|
|
|
|
|
|