|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H3C-CHFBr |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bromine |
|
|
|
Nuclear
Quadrupole Coupling Constants |
|
|
in 1-Bromo-1-Fluoroethane
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bromine nqcc's in
1-bromo-1-fluoroethane were determined by Tatamitani, et al. [1].
Calculation of the nqcc's was made here on the ro/ropt
structure of Ref. [1], and on an approximate equilibrium structure (~ re) derived by MP2/aug-cc-pVTZ optimization with empirically corrected bond lengths, as described here.
Calculated
nqcc's are compared with the experimental values in Tables 1 and 2.
Structure parameters are given in Table 3, atomic coordinates in
Table 4, and rotational constants in
Table 5.
|
|
|
|
|
|
|
|
|
|
|
|
|
In Tables 1 and 2, subscripts a,b,c refer to the
principal axes of the inertia tensor; x,y,z to the principal axes
of the nqcc tensor.
Ø (degrees) is the angle between its subscripted
parameters. ETA = (Xxx - Xyy)/Xzz. |
|
|
|
|
|
|
|
|
|
|
|
|
RMS is the root mean square
difference between calculated and experimental diagonal nqcc's
(percentage of the average of the magnitudes of the experimental
nqcc's). RSD is the calibration residual standard deviation of
the B1LYP/TZV(3df,3p) model for calculation of the bromine nqcc's. |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Table 1. 79Br
nqcc's in BrFHC-CH3 (MHz). Calculation was made on (1) the ro/ropt structure [1], and (2) the ~ re structure. |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Calc. (1)
|
|
Calc. (2) |
|
Expt. [1] |
|
| |
|
|
|
|
|
|
|
|
|
Xaa |
|
499.40 |
|
492.23 |
|
493.49(29) |
|
Xbb |
- |
268.07 |
- |
265.72 |
- |
266.19 ** |
|
Xcc |
- |
231.33 |
- |
226.50 |
- |
227.30 ** |
|
Xab * |
|
0.75 |
|
2.84 |
|
161.8(28) # |
|
|
Xac * |
|
181.22 |
|
180.57 |
|
|
|
|
Xbc * |
|
12.57 |
|
11.80 |
|
|
|
|
|
|
|
|
|
|
|
|
|
RMS |
|
4.27 (1.3 %) |
0.90 (0.27 %) |
|
|
|
RSD |
|
1.58 (0.39 %) |
|
1.58 (0.39 %) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Xxx |
- |
258.54 |
- |
256.56 |
- |
261.95 ## |
|
|
Xyy |
- |
283.35 |
- |
278.51 |
- |
266.19 |
|
|
Xzz |
|
541.89 |
|
535.08 |
|
528.14 |
|
|
ETA |
|
0.0458 |
|
0.0410 |
|
0.0080 |
|
|
Øz,CBr |
|
0.44 |
|
0.82 |
|
0.69 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* The algebraic signs of the
off-diagonal components correspond to the orientation of the molecule
with respect to a,b,c axes given in Table 4. The product XabXacXbc is positive. |
|
|
** Calculated here from the experimental Xaa and Xbb - Xcc = - 38.89(11) MHz. |
|
|
# Absolute value. |
|
|
## Calculated here with Kisiel's QDIAG.f [2], assuming Xab = Xbc = 0, and Xac = 161.8 MHz. Øz,CBr = 0.69o is on the ro/ropt structure. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Table 2. 81Br
nqcc's in BrFHC-CH3 (MHz). Calculation was made on (1) the ro/ropt structure [1], and (2) the ~ re structure. |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Calc. (1)
|
|
Calc. (2) |
|
Expt. [1] |
|
| |
|
|
|
|
|
|
|
|
|
Xaa |
|
417.30 |
|
411.30 |
|
412.42(27) |
|
|
Xbb |
- |
223.97 |
- |
222.00 |
- |
222.49 ** |
|
|
Xcc |
- |
193.33 |
- |
189.30 |
- |
189.93 ** |
|
|
Xab * |
|
0.73 |
|
2.48 |
|
133.31(31) # |
|
|
Xac * |
|
151.28 |
|
150.74 |
|
|
|
|
Xbc * |
|
10.53 |
|
9.88 |
|
|
|
|
|
|
|
|
|
|
|
|
|
RMS |
|
3.53 (1.3 %) |
0.79 (0.29 %) |
|
|
|
RSD |
|
1.38 (0.40 %) |
|
1.38 (0.40 %) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* The algebraic signs of the
off-diagonal components correspond to the orientation of the molecule
with respect to a,b,c axes given in Table 4. The product XabXacXbc is positive. |
|
|
** Calculated here from the experimental Xaa and Xbb - Xcc = - 32.56(11) MHz. |
|
|
# Absolute value. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecular Structure
|
|
|
|
|
|
|
|
|
|
|
|
|
The ro/ropt structure
of Tatamitani et al. consists of bond angles optimized at the
MP2/6-311G(d,p) level of theory, and bond lengths then fitted with
these angles to reproduce the observed moments of inertia. |
|
|
|
|
|
|
|
|
|
|
|
|
The ~ re structure
was calculated by MP2/aug-cc-pVTZ optimization with empirically corrected bond lengths, as described here. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Table 3. BrFHC-CH3 Structure Parameters (Å
and degrees). |
| |
|
|
|
|
| Point Group, C1. |
|
ro/ropt |
~ re |
|
|
|
|
|
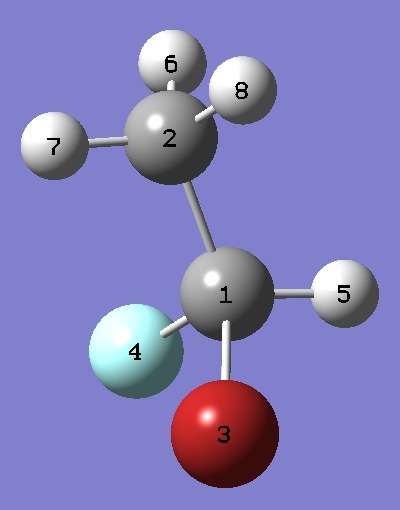 |
C-C |
1.517 |
1.4996 |
| CBr |
1.941 |
1.9436 |
| CF |
1.368 |
1.3443 |
| CH |
1.094 |
1.0863 |
| CH(6) |
1.087 |
1.0897 |
| CH(7) |
1.081 |
1.0872 |
| CH(8) |
1.087 |
1.0874 |
| CCBr |
111.1 |
110.88 |
| BrCF |
108.8 |
108.46 |
| CCH |
113.3 |
113.78 |
| CCH(6) |
108.6 |
108.93 |
|
|
CCH(7) |
110.4 |
109.97 |
| Dihedral angles? See here. |
CCH(8) |
109.8 |
109.88 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Table 4. BrFHC-CH3 Atomic coordinates, ~ re. Normal Species. |
| (More figures are shown than are significant.) |
| |
|
|
|
|
|
|
|
|
|
|
a (Å) |
|
b (Å) |
|
c (Å) |
|
|
|
|
|
|
|
|
|
C |
|
1.0204 |
|
0.0130 |
|
0.4055 |
|
C |
|
1.6871 |
|
1.2448 |
- |
0.1302 |
|
Br |
- |
0.8763 |
|
0.0117 |
- |
0.0186 |
|
F |
|
1.5781 |
- |
1.0846 |
- |
0.1344 |
|
H |
|
1.0754 |
- |
0.0799 |
|
1.4864 |
|
H |
|
2.7488 |
|
1.2061 |
|
0.1124 |
|
H |
|
1.2465 |
|
2.1310 |
|
0.3199 |
|
H |
|
1.5642 |
|
1.2922 |
- |
1.2096 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Table 5. BrFHC-CH3 Rotational Constants (MHz). Normal Species. |
|
|
|
|
|
|
|
ro/ropt |
~ re |
Expt. [1] |
|
|
|
|
|
|
A |
8982.6 |
9180.3 |
8979.428(5) |
|
B |
2883.6 |
2908.6 |
2883.898(3) |
|
C |
2311.4 |
2338.9 |
2310.535(3) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[1] Y.Tatamitani, S.Kuwano, K.Fuchigami, S.Oe, and T.Ogata, J.Mol.Spectrosc. 196,189(1999). |
|
|
[2] Z.Kisiel, PROSPE - Programs for ROtational SPEctroscopy, http://info.ifpan.edu.pl/~kisiel/prospe.htm. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3Br |
CH3CH2Br |
CHBrF2 |
CH2BrF |
|
|
CH2Br2 |
HCCBr |
BrCN
|
CF3Br |
|
|
ClFHC-CH3 |
CH2BrCl
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table of Contents |
|
|
|
|
|
Molecules/Bromine |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BrFHCCH3.html |
|
|
|
|
|
|
Last
Modified 25 Feb 2007 |
|
|
|
|
|
|
|
|
|
|